Our lab uses a combined bioinformatic/experimental approach to investigate the genetic basis and functional consequences of population-specific immune gene splicing and virus-host interaction mechanisms. Addressing such questions as 1) why some immune genes express and splice differently in different human populations or different species? 2) How do these differences play a role in the interaction between immune system and viruses or influence viral host range?
Human Immunodeficiency Virus and Simian Immunodeficiency Virus
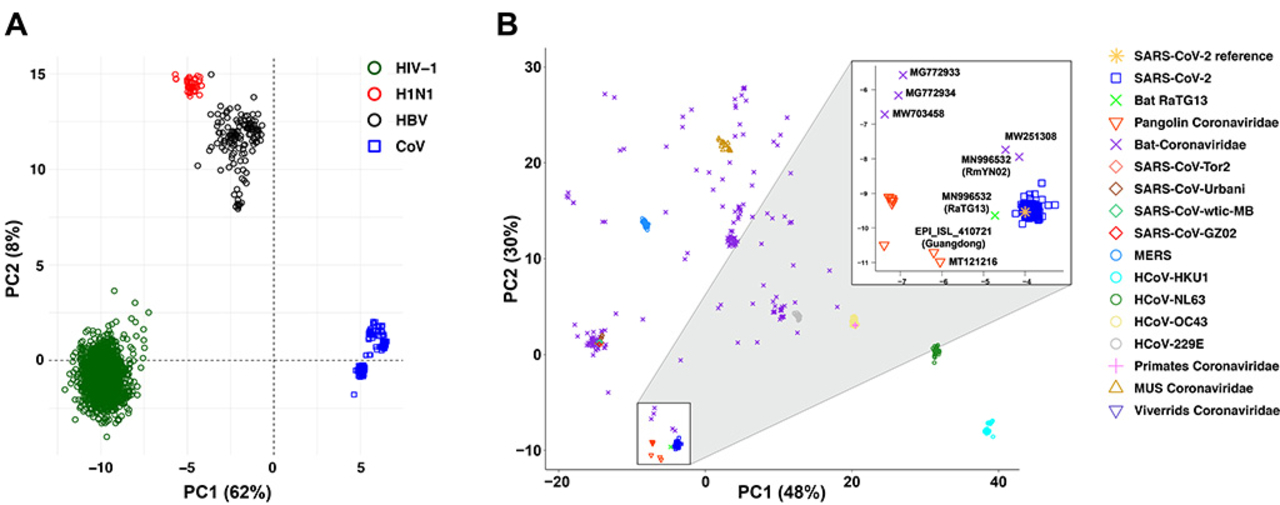
According to UNAIDS, over 36 million people including 1.8 million children under 15 yrs old were infected by HIV in 2017. The rate of new infection worldwide was estimated to be around 5,000 per day, and most of the new infections were in sub-Saharan Africa. HIV infection and its associated illnesses such as tuberculosis, lymphomas, Kaposi sarcoma, and neurocognitive disorder are complex diseases. The immune processes involved in these diseases are numerous and many are yet to be identified and fully characterized. In my lab, data from various sources such as the genome, transcriptome, and proteome of patients and viruses such as HIV are analyzed to determine what viral and host immune factors play a role in our immunity against viruses. Our multi-omic analysis has revealed that some immune genes such as APOBEC3 (A3) undergo differential splicing in different human populations, and this may be one of the reasons for our differential immunity against viruses.
Sars-CoV-2
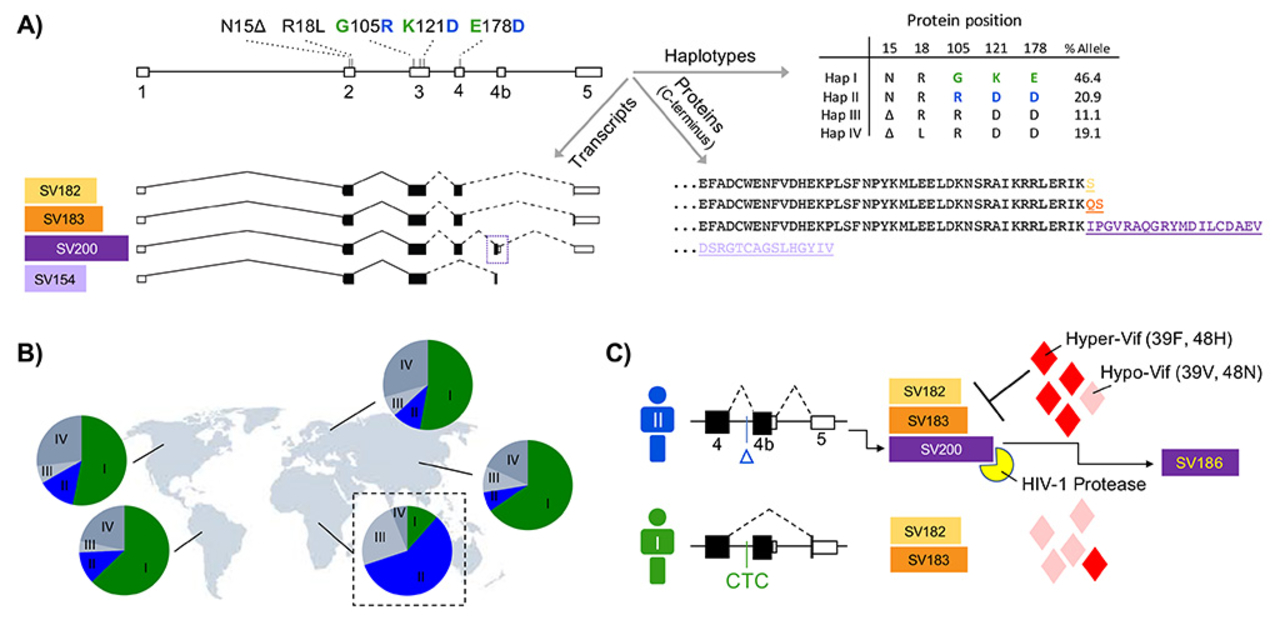
The zinc finger antiviral protein (ZAP) is known to restrict viral replication by binding to the CpG rich regions of viral RNA, and subsequently inducing viral RNA degradation. This enzyme has recently been shown to be capable of restricting SARS-CoV-2. These data have led to the hypothesis that the low abundance of CpG in the SARS-CoV-2 genome is due to an evolutionary pressure exerted by the host ZAP. To investigate this hypothesis, we performed a detailed analysis of many coronavirus sequences and ZAP RNA binding preference data.