Research
Malaria Research Projects
Drug resistance
Artemisinin resistance: Our laboratory first determined that the “slow parasite clearance” observed in Cambodia was due to parasite genetics. We showed that the genes responsible for slow clearance were increasing in frequency along the Thai-Myanmar border and subsequently located the genomic region responsible for much of this variation. Following the identification of Kelch13 in this genomic region, we have documented the dynamics of soft selective sweeps at this locus along the Thailand-Myanmar border.
Representative publications from the lab:
- Anderson TJ et al. High heritability of malaria parasite clearance rate indicates a genetic basis for artemisinin resistance in western Cambodia. J Infect Dis. 2010 May; 201(9):1326-1330. PMC2853733.
- Phyo AP et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: A longitudinal study. Lancet. 2012 May;379(9830):1960-1966. PMC3525980.
- Cheeseman IH et al. A major genome region underlying artemisinin resistance in malaria. Science. 2012 Apr;336(6077):79-82. PMC3355473.
- Anderson TJ et al. Population parameters underlying an ongoing soft sweep in Southeast Asian malaria parasites. Mol Biol Evol. 2017 Jan;34(1):131-144. PMCID: PMC5216669.
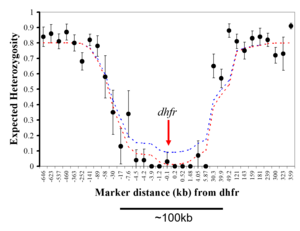
Dynamics of drug resistance evolution: Drug resistance evolution provides a valuable model for understanding adaptation in nature and has significant implications for public health policy. Our group demonstrated that high-level pyrimethamine resistance originated from a single source in Southeast Asia, with a selective sweep around the dihydrofolate reductase (dhfr) gene leading to the loss of genetic variation on chromosome 4. The subsequent transfer of pyrimethamine resistance alleles from Asia to Africa resulted in a significant decrease in the efficacy of antifolate drugs. We have also investigated the role of copy number variation (CNV) in this resistance phenotype, revealing multiple independent origins of copy number amplification in an ABC transporter and demonstrating adaptive changes in CNV during the evolution of antifolate resistance.
Representative publications from the lab:
- Nair S et al. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol. 2003 Sep;20(9):1526-1536. PMID: 12832643.
- Roper C et al. Intercontinental spread of pyrimethamine-resistant malaria. Science. 2004 Aug; 305(5687):1124. PMID: 15326348.
- Nair S et al. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol Biol Evol. 2007 Feb;24(2):562-573. PMID: 17124182.
- Alfred Amambua-Ngwa et al. Chloroquine resistance evolution in Plasmodium falciparum is mediated by the putative amino acid transporter AAT1. Nat Microbiol. 2023 Jul;8(7):1213-1226. PMID: 37169919.
Population genomics
Genetic structure of parasite populations: Our initial work on malaria

developed and utilized microsatellite markers to characterize Plasmodium falciparum populations, revealing dramatic differences in population structure that correlate closely with transmission intensity. We recently observed similar changes in genetic structure within a single population where transmission was declining. Our lab has focused on the genetic composition of multiple-clone infections, demonstrating that these infections consist of related parasite haplotypes, which contradicts simpler models of parasite superinfection. This finding was recently confirmed using FACS-based single-cell genomics methods developed in our laboratory.
Representative publications from the Lab:
- Anderson TJ et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol. 2000 Oct;17(10):1467-82. PMID: 11018154.
- Nkhoma SC et al. Close kinship within multiple-genotype malaria parasite infections. Proc Biol Sci. 2012 Jul; 279(1738):2589-98. PMC3350702.
- Nkhoma SC et al. Population genetic correlates of declining transmission in a human pathogen. Mol Ecol. 2013 Jan;22(2):273-85. PMC3537863.
- Nair S et al. Single-cell genomics for dissection of complex malaria infections. Genome Res. 2014 Jun;24(6):1028-1038. PMC4032849.
Structural variation in parasite DNA: Large-scale deletions, copy number variations (CNVs), insertions, and rearrangements are common in pathogen genomes exhibit high mutation rates, and may underpin rapid adaptation to drugs and other selective pressures. However, structural variants are more challenging to score than SNPs or Indels and have received less attention. We have investigated the role of CNVs in drug resistance evolution and compensatory mutations, as well as the population genomics of CNVs in Plasmodium populations worldwide. We are also using Nanopore long-read sequencing to provide parent-specific references for P. falciparum genetic crosses conducted in our lab. We anticipate that this approach will enhance our ability to identify segregating variants across complex genomic regions that are difficult to access using short-read Illumina sequencing alone.
Representative publications from the Lab:
- Nair S et al. Adaptive copy number evolution in malaria parasites. PLoS Genet. 2008 Oct;4(10):e1000243. PMID: 1897487.
- Cheeseman IH et al. Population Structure Shapes Copy Number Variation in Malaria Parasites. Mol Biol Evol. 2016 Mar;33(3):603-20. PMID: 26613787.

Genomic epidemiology during malaria elimination in Myanmar: We are currently generating and using genomic data from P. falciparum parasites collected from infected patients on the Thai-Myanmar border to better understand malaria transmission during intensive malaria control efforts. This work is conducted in collaboration with Dr. François Nosten (Shoklo Malaria Research Unit) and Dr. Daniel Parker.
Representative publications from the Lab:
Li et al. Impact of intensive control on malaria population genomics under elimination settings in Southeast Asia. Nature Miccrobiology. 2026 April; 11, pages1361–1373. Featured in Clear Sky Science.
Linkage mapping of parasite fitness traits
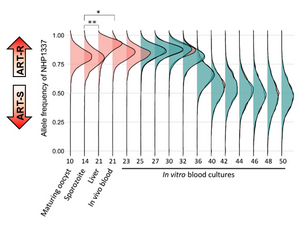
Through a P01 NIH grant, we have been collaborating with Dr. Ashley Vaughan (Seattle Children’s Hospital), Dr. Mike Ferdig (University of Notre Dame), and Dr. Ian Cheeseman (Texas Biomedical Research Institute) to utilize humanized mice for conducting genetic crosses in P. falciparum. This collaboration has enabled us to generate crosses with large numbers of progeny, providing excellent statistical power to localize genes underlying important parasite phenotypic traits. Specifically, we have focused on developing efficient bulk-segregant approaches for analyzing genetic crosses, which do not require laborious cloning and phenotyping of individual progeny. We have applied these methods to identify genomic regions associated with parasite fitness and nutrient acquisition/metabolism.
Representative publications from the lab:
- Vaughan AM et al. Plasmodium falciparum genetic crosses in a humanized mouse model. Nat Methods. 2015 Jul;12(7):631-3. PMC4547688.
- Button-Simons KA et al. The power and promise of genetic mapping from Plasmodium falciparum crosses utilizing human liver-chimeric mice. Commun Biol. 2021 Jun;4(1):734. PMCID: PMC8203791.
- Li X et al. Genetic mapping of fitness determinants across the malaria parasite Plasmodium falciparum life cycle. PLoS Genet. 2019 Oct; 15(10):e1008453. PMCID: PMC6821138.
- Kumar S et al. A Malaria parasite cross reveals genetic determinants of Plasmodium falciparum growth in different culture media. Front Cell Infect Microbiol. 2022; 12: 878496. PMCID: PMC9197316.
Using CRISPR/Cas9 to decipher fitness costs and compensatory mutation evolution
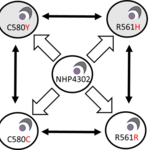
Fitness costs, compensation and epistasis: Drug resistance genes often interfere with key metabolic processes, leading to significant fitness costs for the parasites. We anticipate that compensatory changes will evolve to restore fitness in drug-resistant parasites. Different genes involved in drug resistance evolution may interact in additive or epistatic ways to influence both drug resistance and parasite fitness. We are employing various approaches to identify and understand fitness costs and compensatory evolution in malaria, and to determine whether gene interactions are additive or epistatic in shaping outcome phenotypes. Specifically, we are using experimental evolution, longitudinal population genomics surveys, and CRISPR/Cas9 techniques to gain insights into the roles of fitness, compensation, and epistasis in drug resistance evolution.
Representative publications from the lab:
- Nair S et al. Fitness costs and the rapid spread of kelch13-C580Y substitutions conferring artemisinin resistance. Antimicrob Agents Chemother. 2018 Aug 27;62(9):e00605-18. PMID: 29914963.
Deletions of Histidine Rich proteins and diagnostic evasion in malaria parasites: Histidine-rich proteins are produced in large quantities by P. falciparum and form the basis of rapid diagnostic tests for malaria. However, some parasites carry deletions in the genes encoding these proteins. We are investigating the costs associated with these gene deletions and the compensatory mutations that may emerge to counteract the resulting fitness costs in the parasites.
Representative publications from the lab:
- Nair S et al. Fitness costs of pfhrp2 and pfhrp3 deletions underlying diagnostic evasion in malaria parasites. J Infect Dis. 2022 Nov 1;226(9):1637-1645. PMID: 35709327.
Schistosome Research Projects
Genetic basis of drug resistance

Our lab pioneers the use of laboratory genetic crosses combined with linkage analysis, bulk-segregant analysis, and genome-wide association studies to map the genetic bases of oxamniquine and praziquantel resistance in schistosome parasites. Using these genetic and genomic approaches, we have deciphered the mechanism of action of oxamniquine, a drug used in South America to treat infections with Schistosoma mansoni.
Praziquantel (PZQ) is currently the only drug available to treat schistosomiasis, but resistance could occur in the field. Using a genome-wide association study (GWAS) with a lab population of parasites polymorphic for PZQ resistance, coupled with a new high-throughput phenotyping method based on worm metabolism, we have pinpointed a genomic region involved in PZQ response. This discovery allows us to identify a candidate gene (TRPM.PZQ) involved in PZQ's mode of action, which was previously unknown. Deciphering the PZQ response gene(s) will enable us to: (i) identify genetic markers linked to drug resistance or reduced efficacy, and (ii) develop molecular surveillance approaches to detect drug-resistant parasites in the field, assisting current mass drug administration programs in Afri

ca and adapting treatments.
We are currently collaborating with Dr. Eric Ndombi and his team in Kisumu, Western Kenya, Dr. Yves-Nathan Tian-Bi in Côte d’Ivoire, Dr. Kader Kondé in Guinea, Dr. Steffi Knopp, Dr. Mohammed Said, and Dr. Humphrey Mazigo in Zanzibar Tanzania to investigate the genetic diversity of the TRPM.PZQ gene in field-collected samples of S. mansoni and S. haematobium. Functional validation of any mutations found in the field will be analyzed for PZQ resistance/susceptibility phenotypes through our collaboration with Prof. Jonathan Marchant at the Medical College of Wisconsin.
Representative publications from the Lab:
- Valentim CL et al. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science. 2013 Dec;342(6164):1385-1389. PMC4136436.
- Chevalier FD et al. Independent origins of loss-of-function mutations conferring oxamniquine resistance in a Brazilian schistosome population. Int J Parasitol. 2016 Jun;46(7):417-24. PMID: 27073078.
- Le Clec'h W, Chevalier FD et al. Genetic analysis of praziquantel response in schistosome parasites implicates a transient receptor potential channel. Sci Transl Med. 2021;13(625):eabj9114. PubMed PMID: 34936381.
- Chevalier FD, Le Clec'h W et al. A single locus determines praziquantel response in Schistosoma mansoni. Antimicrob Agents Chemother. 2024 Mar 6;68(3):e0143223. PMID: 38289079.
Genetic basis of transmission traits
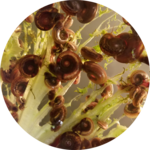
Host-specificity: Host-pathogen compatibility is determined by both host and parasite genes. There are multiple models of how this evolves (gene-for-gene coevolution and gene matching models), but there are few examples where this is understood at the molecular level. We are using genetic crosses and bulk-segregant approaches to determine the Schistosome parasite genes involved in compatibility with their snail hosts. On the other side of the interaction, the Blouin lab is using similar genetic approaches to identify the snail genes involved in compatibility and resistance to schistosome parasites. We hope to understand the molecules involved in these compatibility and resistance mechanisms in both the snail and the schistosome parasite.
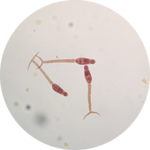
Cercarial production and chronobiology: We have characterized striking differences in virulence, transmission, and parasite growth dynamics between two Schistosoma mansoni populations. One population of parasites is particularly virulent for snail and mammal hosts, linked to significant production of sporocysts and cercariae in the snail host and eggs in the mammal host, while the other exhibits a low virulence phenotype. By conducting genetic crosses between parasites from these two populations, we have revealed that cercarial production involves multiple genes. Identifying the causative genes will require a better understanding of sporocyst biology and the different cell types that constitute them.
Cercarial production is not continuous but rather rhythmic and dependent on the

time of day. For example, some schistosome parasites shed cercariae during daylight, while others shed cercariae at night. Using genetic crosses and classical linkage mapping between populations of diurnal and nocturnal S. mansoni, we aim to decipher the genetic basis of these chronobiological adaptive traits. Our work on cercarial chronobiology is done in collaboration with Dr. Hélène Moné and Dr. Gabriel Mouahid from the University of Perpignan in France.
Understanding how parasites adapt and develop within their hosts is crucial for identifying new therapeutic targets, leading to better control of schistosome parasites and, eventually, the schistosomiasis disease.
Representative publications from the Lab:
- Le Clec'h W et al. Striking differences in virulence, transmission and sporocyst growth dynamics between two schistosome population. Parasit Vectors. 2019 Oct 16;12(1):485. PMID: 31619284.
- Le Clec'h W et al. Genetic architecture of transmission stage production and virulence in schistosome parasites. Virulence. 2021 Dec;12(1):1508-1526. PMID: 34167443.
- Le Clec'h W et al. No evidence for schistosome parasite fitness trade-offs in the intermediate and definitive host. Parasit Vectors. 2023 Apr 17;16(1):132. PMID: 37069704.
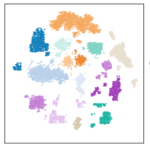
Single-cell transcriptomics of Schistosome sporocysts: Our goal is to develop an atlas of sporocyst cell types and to identify differences in cell composition and gene expression of these cell types that could be linked to cercarial production. This atlas will be a valuable resource for the schistosome research community and could be used to develop cell-type markers for spatial transcriptomic approaches.
Population genomics
Schistosome invasion of the Americas: Using whole genome amplification and sequencing of single miracidia larvae from infected patients preserved on FTA cards, we examined the genomic impact of Schistosoma mansoni colonization in South America during the transatlantic slave trade. We found a significant reduction in genetic diversity and slower decay in linkage disequilibrium (LD) in parasites from East to West Africa. Despite this, nuclear diversity and LD in West Africa and Brazil were similar, indicating minimal bottlenecks and barriers to colonization. Our data imply that unsampled populations from central Africa, including contributions from Niger, were likely the main sources of Brazilian S. mansoni. The lack of a bottleneck suggests that this invasion was likely facilitated by pre-adapted parasites that established themselves relatively easily in the Americas.
Representative publications from the Lab:
- Platt RN et al. Genomic analysis of a parasite invasion: Colonization of the Americas by the blood fluke Schistosoma mansoni. Mol Ecol. 2022 Apr;31(8):2242-2263. doi: 10.1111/mec.16395. Epub 2022 Feb 25. PMID: 35152493.
Schistosome hybridization: Schistosoma haemat

obium, which causes urinary schistosomiasis in humans, and the related livestock parasite Schistosoma bovis can interbreed in the laboratory and produce fertile offspring. Previous studies have suggested ongoing hybridization between these species and others in the S. haematobium clade, though they used limited genetic markers. Using whole genome amplification and exome/genome sequencing of single S. haematobium miracidia larvae preserved on FTA cards from infected patients in West and East Africa, we found no evidence of contemporary hybridization between S. bovis and S. haematobium. However, all S. haematobium genomes from West Africa showed hybrid ancestry, with 3.3-8.2% of their nuclear genomes derived from S. bovis, indicating an ancient introgression event. We are now combining population genomics and laboratory genetic crosses to further investigate the genomic consequences, frequency, and phenotypic effects of this introgression.
Representative publications from the Lab:
- Platt RN et al. Ancient Hybridization and Adaptive Introgression of an Invadolysin Gene in Schistosome Parasites. Mol Biol Evol. 2019 Oct ;36(10):2127-2142. PMC6759076.

Long read sequencing to maximize the utility of parasite genetic crosses: Schistosome genomes are rich in transposable elements (TEs) and structural rearrangements, which are challenging to resolve with Illumina short-read sequencing alone. We are using Nanopore long-read sequencing to generate parent-specific reference sequences for the schistosome genetic crosses conducted in our lab. We anticipate that this approach will enhance our ability to align reads across the entire genome and identify TE insertions and structural variants that may be driving quantitative trait loci (QTLs) linked to several phenotypes of interest that we are investigating.
The snail-microbiome-schistosome interaction
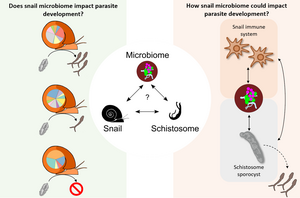
Dr. Frédéric Chevalier and Dr. Winka Le Clec’h, staff scientists in the Anderson lab, are co-leading a schistosome-snail-host-microbiome project. They investigate the diversity and abundance of microorganisms within the hemolymph (i.e., blood) and organs of Biomphalaria snails. They have highlighted significant differences in microbiome composition at the level of individual snails, snail populations, and species. They have also established a snail organs microbiome atlas and are currently investigating the impact of schistosome infections on the snail microbiome with NIH R21 support. Understanding the role of the microbiome in the snail-parasite interaction can lead to better control of parasite populations, especially if specific bacteria help snails resist infection.
Representative publications from the Lab:
- Chevalier FD et al. The hemolymph of Biomphalaria snail vectors of schistosomiasis supports a diverse microbiome. Environ Microbiol. 2020 Dec;22(12):5450-5466. PMID: 33169917.
- Le Clec'h W et al. Snails, microbiomes, and schistosomes: a three-way interaction? Trends Parasitol. 2022 May;38(5):353-355. PMID: 35190282.
- Carruthers LV et al. How should we sample snail microbiomes? bioRxiv [Preprint]. 2024 Jun 11:2024.06.11.598555. PMID: 38915569.
Main technologies and methods used:
Parasite lifecycles: In our laboratory, we actively maintain the full

lifecycle of several populations of schistosome parasites (S. mansoni and S. haematobium) using our snail colonies and rodents (hamsters or mice). We currently have inbred and outbred snail populations of Biomphalaria glabrata from Brazil, B. alexandrina from Egypt, B. sudanica from Kenya, Bulinus truncatus, and Bulinus globosus from Kenya. Additionally, we have a diverse collection of cryopreserved Plasmodium falciparum parasites (from various African and Southeast Asian countries), which we can propagate asexually using human blood.

Molecular biology and cell biology: In our laboratory, we use a wide variety of molecular biology and cell biology techniques, which we have developed and/or adapted for our non-model organisms of interest: Plasmodium falciparum and Schistosoma mansoni. These techniques include:
- Library preparation for high-throughput sequencing (Whole Genome sequencing, Exome capture, RNAseq, MiSeq, Nanopore Long-read sequencing).
- Targeted-capture array
- Whole genome amplification (from FTA card preserved samples or gDNA)

- PCR / Nested PCR / quantitative PCR / RT-PCR and qRT-PCR / PCR-RFLP
- In-vitro culture and assay for P. falciparum and Schistosome worms
- Single-cell RNAseq for Malaria parasites and Schistosome sporocysts
- CRISPR gene editing
- Phenoloxidase activity dosage in snail blood
- Drug treatment assay (for both Malaria and Schistosome worms)

Data analysis: All data generated in our lab are analyzed in-house using custom pipelines (Conda environments, etc.) and scripts (Bash, R, Python, etc.) on the Texas Biomed High-Performance Computing Cluster. These analyses include:
- WGS, Exome, Microbiome, RNAseq, scRNAseq pipelines for Malaria and Schistosome parasites
- Quantitative Trait Loci (QTL) analysis
- Bulk-segregant analysis
- GWAS analysis
- Phylogenetic analysis
- Population genomics
- Admixture analysis
- Statistical analysis using R (survival analysis, multivariate analysis etc.)